GABA
GABA is the major inhibitory amino acid transmitter of the mammalian central nervous system and it is present in some 40% of all neurones.
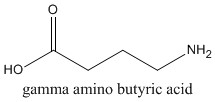
Synthesis, storage and function
Gamma amino butyric acid (GABA) is the primary inhibitory neurotransmitter in the central nervous system. It is found in almost every region of brain, and is formed through the activity of the enzyme glutamic acid decarboxylase (GAD).

GAD catalyzes the formation of GABA from glutamic acid. The synthesis of GABA is linked to the Kreb's cycle. GAD requires vitamin B6 (pyridoxal phosphate) as a cofactor, which can be used to regulate the levels of GABA. GABA is destroyed by a transamination reaction, in which the amino group is transferred to alpha-oxoglutaric acid (to yield glutamate), with the production of succinic semialdehyde, and then succinic acid. The reaction is catalysed by GABA transaminase.
Vigabatrine, a GABA agonist, is used to treat epilepsy by inhibiting GABA transaminase.
GABA-ergic neurons and astrocytes take up GABA by specific transporters, and it is this, rather than GABA transaminase, that removes the GABA after it has been released. GABA transport is inhibited by Guvacine and Nipecotic acid.
Compounds such as the competitive GAD inhibitor allylglycine, inhibit GABA formation and cause convulsions due to the lack of GABA activity.

Sodium valproate (or valproic acid) on the other hand, blocks GABA transaminase activity, thereby elevating GABA levels, and thus alleviating seizures. Sodium valproate is useful in the treatment of epilepsy and bipolar mood disorders. Another strategy is to block GABA-transaminase with g-acetylenic GABA, thereby increasing the concentration of GABA at the synapse.
GABA receptors
Structure and Pharmacology
The GABA receptors are a class of receptors that respond to the neurotransmitter gamma aminobutyric acid (GABA), the chief inhibitory neurotransmitter in the vertebrate central nervous system. There are three classes of GABA receptors: GABAA, GABAB, and GABAС.
GABAA and GABAС receptors are ligand-gated ion channels (also known as ionotropic receptors), whereas GABAB receptors are G protein-coupled receptors (also known as metabotropic receptors).
Ligand-gated ion channels
GABAA
It has long been recognized that the fast response of neurons to GABA that is blocked by bicuculline and picrotoxin is due to direct activation of an anion channel.
This channel was subsequently termed the GABAA receptor. Fast-responding GABA receptors are members of family of Cys-loop ligand-gated ion channels. Members of this superfamily, which includes nicotinic acetylcholine receptors, GABAA and GABAС receptors, glycine and 5-HT3 receptors, possess a characteristic loop formed by a disulphide bond between two cysteine residues.
The GABAA receptor consists of three separate subunits with an alpha2, beta2, gamma arrangement similar to that found for neuronal nicotinic receptors. The ligand binding site is located at the interface between the alpha and beta subunits. The benzodiazepine binding site is located at a similar level at the interface between the alpha and gamma subunits. Each subunit is comprised of four hydrophobic sequences which span the membrane, with a large extracellular amino terminus containing the binding site and a carboxyl terminus located intracellularly.

Electrophysiological studies demonstrated that the activation of the receptor resulted in increased chloride conductance of the cell membrane with the concentration-response curve exhibiting positive cooperativity, consistent with the presence of at least two agonist binding sites on the receptor molecule. The agonist induced current decreased on continued exposure to high agonist concentrations suggesting that these receptors undergo desensitisation.
GABAC
In addition to the GABAA receptors there is a distinct class of ligand gated ion channels that are activated by GABA; referred to as the GABAC receptor. The natural agonist GABA is about an order of magnitude more potent at the GABA receptors than at the most common of the GABA receptors. The GABA receptors are activated by cis-aminocrotonic acid (CACA), which is not recognised by either the GABAA or GABAC receptors, suggesting that they recognise the partially folded conformation of GABA.
GABAC receptors are not blocked by bicuculline and do not recognise the benzodiazepines, barbiturates or the neuroactive steroids but, like GABAA receptors are blocked by picrotoxin, while 1,2,5,6-tetrahydropyridine-4-yl methyl phosphinic acid appears to inhibit GABAC receptors selectively.
However, molecular cloning studies have revealed that this pharmacological profile is remarkably similar to that exhibited by the rho-subunits when expressed ectopically. Two homologous subunits, rho1 and rho2, have been identified in man and these can be expressed as homomers or heteromers, but do not co-assemble with any of the GABA receptor subunits.
Common characteristics
In ionotropic GABAA and GABAС receptors, binding of GABA molecules to their binding sites in the extracellular part of receptor triggers opening of a chloride ion-selective pore.
Opening of a chloride conductance drives the membrane potential towards the reversal potential of the Cl¯ ion which is about –80 mV in neurons, inhibiting the firing of new action potentials.
However, there are numerous reports on GABAA receptors, which are actually excitatory. This phenomenon is due to increased intracellular concentration of Cl¯ ions either during development of the nervous system or in certain cell populations.
After this period of development, a Chloride pump is up-regulated and inserted into the cell membrane, pumping Cl- ions into the extracellular space of the cell. Further openings via GABA binding to the receptor then produce inhibitory responses. Over-excitation of this receptor induces receptor remodeling and the eventual invagination of the GABA receptor. As a result, further GABA binding becomes inhibited and IPSPs are no longer relevant.
G protein coupled receptor: GABAB
A slow response to GABA is mediated by GABAB receptors, originally defined on the basis of pharmacological properties.
In studies focused on the control of neurotransmitter release, it was noted that a GABA receptor was responsible for modulating evoked release in a variety of isolated tissue preparations. This ability of GABA to inhibit neurotransmitter release from these preparations was not blocked by bicuculline, was not mimicked by isoguvacine, and was not dependent on Cl¯, all of which are characteristic of the GABAA receptor. The most striking discovery was the finding that baclofen (β-parachlorophenyl GABA), a clinically employed spasmolytic mimicked, in a stereoselective manner, the effect of GABA.
Later ligand-binding studies provided direct evidence of binding sites for baclofen on central neuronal membranes. cDNA cloning confirmed that the GABAB receptor belongs to the family of G-protein coupled receptors. Additional information on GABAB receptors has been reviewed elsewhere.
Drugs acting on GABA receptors
Below is the table showing the effector pathway, agonists and antagonists of GABA receptors.

GABAA
GABA binds to GABAA receptors in the extended form as demonstrated by the activity of trans 4-aminocrotonoic acid and the lower activity of cis 4-aminocrotonoic acid.

Muscimol is a naturally occurring compound isolated from Amanita muscaria and acts as a GABAA agonist. The isoxazole ring also is found in the GABAA agonist tetrahydroisoxazolopyridinol (THIP).

Bicucullin acts as a direct antagonist at the GABAA receptor as does SR 42641.

GABAB
Baclofen is direct agonist at GABAB receptors, which are coupled to G proteins. GABAB receptors may regulate Ca2+ and K+ influx through the Gi/o family of G proteins and act presynaptically to inhibit the release of excitatory amino acids such as glutamate. Baclofen is orally active as a muscle relaxant and has been used in the treatment of rigidity and spasticity of cerebral palsy. Phaclofen is a weak partial agonist, while saclofen is an antagonist at GABAB receptors.
Anxiolytics: Benzodiazepines
Benzodiazepines represent a class of compounds collectively referred to as anxiolytics. Benzodiazepines modulate the binding of GABA to the GABAA receptor. Benzodiazepines increase the binding of GABA to GABAA receptors and promote Cl- influx.
All the overt effects of the benzodiazepines: sedative, anxiolytic, anticonvulsant, muscle relaxant and amnesic, are produced via the GABAA receptors.
Diazepam (Valium) is one of the most widely prescribed sedatives on the market.

Benzodiazepines exhibit anxiolytic activity due to their ability to promote GABA binding. They act as indirect agonists. Several ligands can block the actions of benzodiazepines, including flumazenil, a benzodiazepine antagonist.
Anticonvulsant drugs
Anticonvulsants are used to treat the various forms of epilepsy, which is characterized by excessive neuronal firing in the cortical and temporal lobe regions of the brain. Electroencephalograms (EEG) can pick up the rhythmic discharge of neurons from electrodes placed on the scalp. Rhythmic discharges of spikes and slow waves characterize the EEG during seizures.
The benzodiazepines have an anticonvulsant action in addition to the anxiolytic activity. Recent work has helped distinguish between the two roles for the benzodiazepines. Picrotoxin is a convulsant which interacts with the GABA receptor complex and blocks the Cl- ionophore.
Diphenylhydantoin (phenytoin) is useful in the treatment of epilepsy. Phenytoin stabilizes the neuron against the excitation associated with seizure activity, without producing sedation.









